
β-thalassemia
- β-thalassemia is also termed as erythroblastic anaemia, β-type microcytemia, or Mediterranean anaemia.
- The term thalassemia was coined by George Whipple and William Bradford, in which thalassa depicts sea and -emia meaning blood. Thus, thalassemia refers as ‘sea in the blood’.
But, for Greeks the sea was represented by Mediterranean thus, thalassemia relates the idea of Mediterranean in the blood. - β-thalassemia is one of the most common autosomal recessive diseases in the world.
- It is the inherited blood disorder indicated by the condition where the production of beta globin chains of haemoglobin tetramer (formed of 2-α-chains and 2-β-thalassemia: Causes, types, symptoms, diagnosis, and treatment -chains) is either reduced or absent.
- Haemoglobin is the iron-containing protein in RBCs responsible for carrying oxygen to all parts of the body.
- In case of person with β-thalassemia, the haemoglobin is defective, resulting low concentration of oxygen in blood supply ultimately causing anaemia.
- It is seen most frequently in people of Mediterranean region, the Middle East, Central Asia, India, and the Southeast Asia.
Causes:
- Normally, Haemoglobin is composed of 2 identical a-chains and 2 identical β -chains that are prone to mutations.
- Mutation in β-chains (globin protein) producing gene ie HBB gene causes β-Thalassemia.
- The beta-globin (HBB) gene is mapped in short arm of chromosome no. 11 with 4 other functional globin genes i.e. the delta globin gene, the embryonic epsilon gene, the G-gamma and the fetal A-gamma genes, and a pseudogene (ψB1).
- HBB gene accounts for the formation of beta-globin protein.
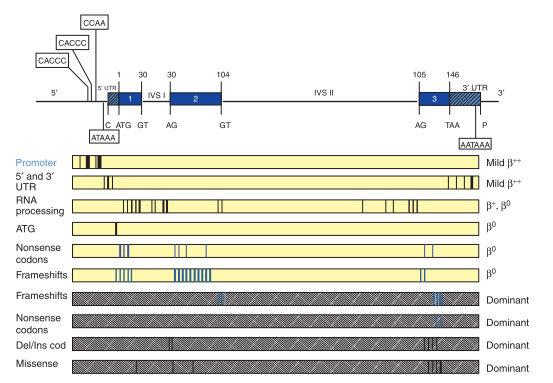
- β-thalassemia being heterogenous at molecular level and prone to mutation. More than 200 disease causing mutations have been recognized till date.
- In most cases, the major mutations such as-substitution of single nucleotide, deletions or additions of oligonucleotides causes β -thalassemia.
- Point mutations altering the expression of the β-globin by one or more of these different mutation:
- Transcriptional mutations on promoter region (CACCC or TATA box) and UTR region: this result in faulty β-gene transcription indicated by reduced production of β-chains, reduced polyadenylation signal and splicing defects.
- Mutations that has impact on messenger RNA (mRNA) processing by change in initiation codon, splicing and frameshift mutations.
- Substitution mutations leading in abnormal globin protein.
- In rare cases, β -thalassemia occurs from gross gene deletion which is indicated by absence of β-chains production.
Types of β-thalassemia:
On the basis of severity of symptoms, b-thalassemia is categorized into two classes:
1) Beta thalassemia major (aka Cooley’s anaemia)
2) Beta thalassemia minor
1. Thalassemia major:
- It is also termed as Cooley’s anaemia.
- If the newly born baby inherits two mutated gene, then it results in this condition and the signs and symptoms are seen to vary from moderate to severe within 2 years of age.
- The child is said to be homozygous for β-thalassemia.
- These infected individuals require regular blood transfusion for survival.
- Symptoms include:
- Paleness
- Splenomegaly results increase in size of abdomen.
- Loss of appetite
- Irritability
- Fatigue
- Retarded growth
- Expansion of bone marrow leading to irregular bone deformities.
- Complications
- It rises for the individuals taking regular blood transfusions after 10-11 years.
- The post transfusional iron overload related complications occur which include slow growth and sexual immaturity, heart related difficulties, liver (cirrhosis, chronic hepatitis), and irregular functioning of endocrine glands.
- Likewise, there is high chance of hepatocellular carcinoma due to iron overload and liver-viral infection.
- Individuals lacking regular blood transfusions often die before third decade.
2. Thalassemia minor:
- If the newly born baby inherits single mutated gene, then it is termed as thalassemia minor and the symptoms seen are few to moderate.
- The child is said to be heterozygous for β-thalassemia.
- No any transfusions or occasional transfusions is required.
- It is further divided as:
- i) thalassemia minima: shows no or few symptoms.
- ii) thalassemia intermedia: causes mild to severe anaemia.
- Symptoms include:
- Pale skin
- Jaundice
- Gallstones formation
- Splenomegaly
- Osteoporosis
- Skeletal deformities
- Thrombotic complications
Diagnosis:
- Diagnosis is performed at the age of 6-12 years of age.
- Incase of pregnant woman, the condition in baby is diagnosed by use of amniocentesis or chorionic villus sampling (CVS).
- Following tests are carried out for the detection of beta-thalassemia:
- 1. Complete blood count (CBC):
- It is done to check the number, size and development of different blood cells in a given volume of blood that discloses microcytic hypochromic anaemia, nucleated RBCs on peripheral blood smear.
- 2. Haemoglobin electrophoresis:
- A lab test, Hemoglobin electrophoresis with hemoglobin F and A2 quantitation is performed to differentiate the types of haemoglobin in the blood, which on analysis reveals reduced amount of HbA and increased amounts of HbF after age 12months along with severity of anaemia.
- 3. Molecular and genetic test: Genetic testing can be proved useful for age under 12 months by identification of biallelic pathogenic variants in HBB gene.
Treatment:
- In thalassemia major, the regular blood transfusions is must, that suppress erythropoiesis and halts the increased gastro-intestinal uptake of iron.
- In thalassemia minor, the treatment is symptomatic and is based on folic acid supplementation and splenectomy.
- After 10-12 transfusions, chelation therapy is initiated to control complications caused by iron load.
- Regular gallbladder echography should be done in order to detect cholelithiasis in early stage.
- Evaluation of liver function tests should be performed.
- MRI techniques are involved for the assessment of heart and liver iron after age 10 years.
- Bone densitometry, liver ultrasound, serum ferritin concentration determination etc should be performed time to time.
References:
- https://www.nature.com/articles/gim201012#Sec23
- https://www.ncbi.nlm.nih.gov/books/NBK1426/
- https://www.medicinenet.com/beta_thalassemia/article.htm
- https://www.mayoclinic.org/diseases-conditions/thalassemia/symptoms-causes/syc-20354995
- https://www.hopkinsmedicine.org/health/conditions-and-diseases/beta-thalassemia