
Thalassemia
- Thalassemia is a common term for a group of inheritable, genetic diseases characterized by reduced levels of Hemoglobin, low RBCs production and anemia.
- There are two main types of thalassemia:
1) α-thalassemia: caused by inactivation or loss of alpha globin gene that result in complete absence or minimized production of α– chain of Hb.
2) β-thalassemia: caused by inactivation or loss of beta globin gene that result in complete absence or decreased production of β– chain of Hb
Alpha-Thalassemia:
- Alpha-Thalassemia arises due to inactivation of alpha globin genes. This leads to comparative increase in non-functional beta globin or gamma globin tetramers and causing cell damage ultimately.
- In people with alpha thalassemia, decrease in the level of hemoglobin is seen that obstructs the oxygen from reaching different body parts.
- It is the common blood disorder across the world. It is often observed in people belonging to Mediterranean region, the Middle East, central Asia, India, Southern China, and Africa.
Causes:
- For the condition of alpha thalassemia, the reduction or absence in production of alpha globin chains is accountable.
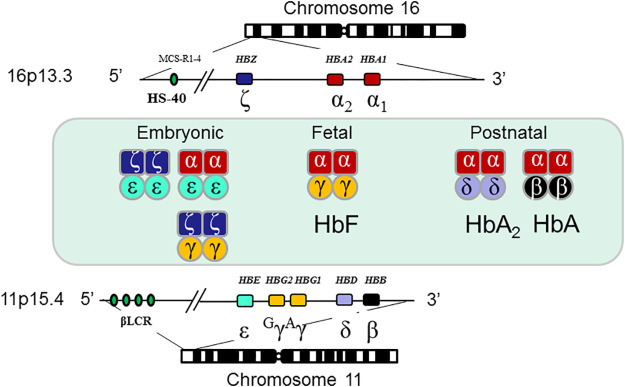
- The reduction of production of alpha globin chains is due to the deletion or mutation of one or more of 4 alpha globin genes (HBA1 and HBA2) situated on short arm of chromosome no.16.
- Individuals possess two copies each of HBA1 gene and HBA2 gene in each cell. Each copy is termed as an allele.
- For each gene, one allele is inherited from an individual’s father and the other from the mother.
- alpha globin production requires all 4 alleles.
- Loss of some or all of these alleles results in the various forms of alpha thalassemia.
- Mutations in the alpha genes are transferred to offspring in an autosomal recessive mode.
Types of alpha-thalassemia:
- Alpha thalassemia are classified on the basis of severity of symptoms:
- Alpha Thalassemia silent carrier
- Alpha Thalassemia Trait
- Haemoglobin H disease
- Haemoglobin H-Constant Spring
- Alpha Thalassemia major
1. Alpha thalassemia silent carrier:
- It is condition where 3 among 4 of the HBA genes are functional and remaining one is mutated.
- This state is confirmed by DNA tests and no any signs and symptoms are prominent in the carrier. The carrier doesn’t face any health problem during the lifetime.
- However, the individuals can transfer the faulty gene onto their offspring.
Alpha thalassemia trait:
- Also termed as alpha thalassemia minor.
- This condition is indicated by the 2 functional genes coding for production of alpha globins.
- The two genes can be present either on same chromosomes or on each of the pair.
- Those genes occurring on same chromosomes are termed as cis-type which is most commonly found in people of Asian descent and those present on each of the pair are termed as trans-type which is prominent in people of African descent.
- The cis-type when co-inherited with another cis-type or haemoglobin H disease yields alpha thalassemia major, or hydrops fetalis.
3. Haemoglobin H disease:
- This condition is depicted by only one functional gene coding for the production of alpha globins, i.e. three of the other genes are non-functional or missing.
4. Haemoglobin H constant spring:
- It is the unusual condition seen in silent carrier state.
- The termination codon mutation results in the elongation alpha globin.
- Generally, children with Hg H constant spring are comparatively more affected than children with classic Hg H disease.
5. Alpha thalassemia major:
- It is also termed as hydrops fetalis.
- It is the most critical form of alpha thalassemia.
- It is indicated by the deletion of all four genes that codes for alpha globin, i.e. all 4 genes are missing.
- In most of the cases, the demise of baby takes place before birth.
Symptoms:
- The characteristic signs and symptoms of the alpha thalassemia condition vary largely from one individual to other.
- People with both alpha thalassemia silent carrier and alpha thalassemia minor do not show any symptoms or alpha thalassemia minor can be mildly anaemic.
- Haemoglobin H disease and alpha thalassemia major shows prominent symptoms.
1) Haemoglobin H disease:
- Symptoms varies from individual to individual.
- The related symptoms are:
- Fatigue
- Pallor
- Weakness
- Loss of appetite
- Difficulty in breathing
- Jaundice
- Headache
- Failure in growth and development of body
- Malnutrition
- Swelling of abdomen due to splenomegaly
- Hepatomegaly
- Leg ulcers
- Gall stones
- Deficiency of folic acid
2) Alpha thalassemia major :
- It is also termed as Haemoglobin (Hb) Bart’s hydrops fetalis, being the most severe form of alpha thalassemia.
- The term hydrops fetalis represents the acquisition of edema (excess amount of fluids) in the tissues and organs of a developing foetus.
- Symptoms are:
- Anaemia
- Hepatomegaly
- Splenomegaly
- Hydrocephaly( unusual collection of cerebrospinal fluid within the skull)
- Heart failure
- Distorted skeleton and urinogenital tract
- poor brain development
Diagnosis:
- Diagnosis in infants takes place through new born screening for Alpha thalassemia.
- In most of cases, diagnosis for Hb Bart’s hydrops fetalis takes place before birth.
- Blood test:
- Suspected individuals are required to undergo blood tests such as Complete blood count (CBC). This test measures the number, size and shape of RBCs and also the concentration of haemoglobin.
- Molecular genetic testing
- It is done for detecting mutations in HBA1 and HBA 2 genes.
- Doppler ultrasonography:
- It is used to measure the blood flow rate through the cerebral arteries in the foetus for detecting Hb Bart’s hydrops fetalis.
- It is a prenatal diagnosis during pregnancy.
Treatment:
- Many people with Hb H disease do not require treatment but in some cases they can be recommended to have folic acid and vitamin supplementation.
- Blood transfusion:
- Some individuals require frequent blood transfusions during early age or later adulthood.
- Individuals requiring regular blood transfusions may have complications due to iron overload. This iron overload condition can be treated by chelation therapy in which deferoxamine is used as iron chelator and allows iron to dissolve in water and is excreted out. Deferiprone and deferasirox are also used to reduce level of iron.
- Splenectomy:
- It is advised for the individual with massive spleen enlargement or hypersplenism condition.
- Cholecystectomy:
- It is also recommended for the removal of gall bladder incase of frequent occurrence of gallstones.
- Proper nutrition:
- Nutrition advices can be taken from dietician for the proper diet.
- Gene therapy
- It is also being studied for the treatment where the defective gene is replaced with normal gene, which is supposed to cure the disease.
Reference:
- https://rarediseases.org/rare-diseases/alpha-thalassemia/
- https://thalassemia.com/what-is-thal-alpha.aspx#gsc.tab=0
- https://www.hopkinsmedicine.org/health/conditions-and-diseases/alpha-thalassemia
- https://ghr.nlm.nih.gov/condition/alpha-thalassemia#synonyms